
Project information
- Category: Genome assembly and annotation
- Coordination: Yann Guiguen
- Project date: 2018 - 2021
Evolution of genes and genomes after whole genome duplication
The sigenae team was in charge of the de novo assembly and annotation of numerous fish genomes.
Genome assembly
Depending on biological resources availability and of DNA extraction feasibility for different fish
species, genomes were assembled by mixing different sequencing data types. Most genomes were assembled
from long read ONT (Oxford Nanopore Technology) sequences and Hi-C read Illumina sequences.
For some species, additional Illumina reads were produced by a 10x Genomics protocol.
For some more recently sequenced species, ONT reads were replaced by PacBio HiFi reads.
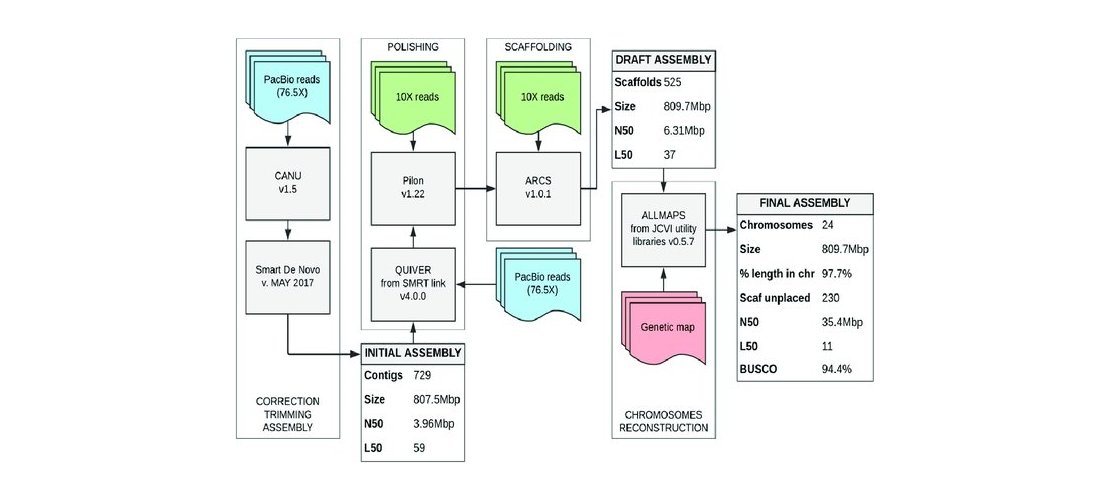
The protocol to asssemble ONT reads included read trimming, size-filtering, assembling and polishing
using long reads then using short reads steps. The HiFi reads assembly protocol includes only an
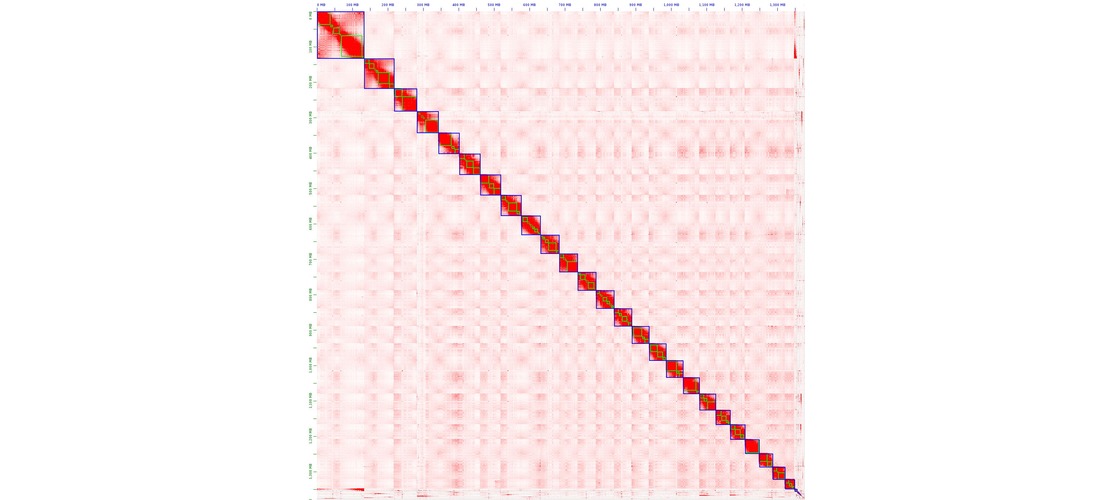
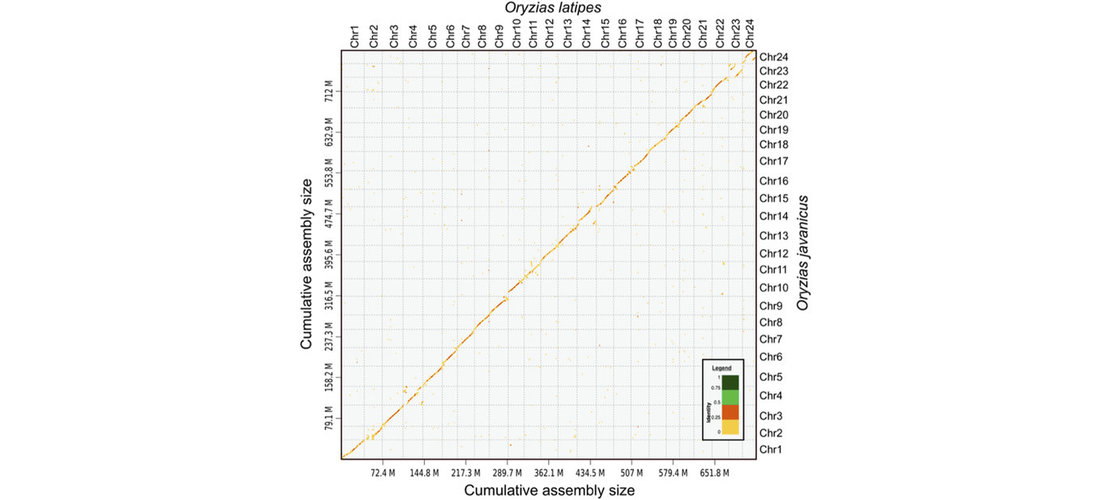
assembly step. Contigs produced by the assembly were then scaffolded to reach chromosome scale
assemblies. The strategy used was to generate Hi-C reads, to map those reads on the contigs and
perform an assembly scaffolding step. Then scaffolds were manually reviewed to correct common assembly
errors as misjoins, translocations or inversions.
Genome annotation
Genome annotation was done by an in-house pipeline. The first step was identifying repetitive content.
Species specific de novo repeats library were built and used together with reference repeat library to
locate repeated regions before soft-masking them. The next step was devoted to collect transcription
evidences in order to perform ab initio gene prediction. Evidences come from similarity search with
known fish proteins, assembled transcripts from RNA-Seq data and hints from orthologous gene location.
Evidences and predictions are next combined to determine the best set of gene models. Finally, the
genome annotation gene completeness was assessed by comparison with close species orthologous gene sets.