Support information
- Category: Genome assembly
- Coordination: support.sigenae@inrae.fr
- Service: since 2018
- Project example: GenoFish
Genome assembly
Sequencing technology evolution toward long high quality reads enables today to assemble large genomes
(1Gb and more) for a few thousand euros. Which makes it possible for a team or even a single researcher
to produce the reference best adapted for her/his daily work. Genome assembly is know to be like
building a jigsaw puzzle, the larger the pieces the easier the work. Long read assembly will produce
contigs which depending on the read quality will have to be corrected (Nanopore or PacBio CLR) or not
(PacBio HiFi). Contig correction is performed by aligning long and/or short reads and updating bases in
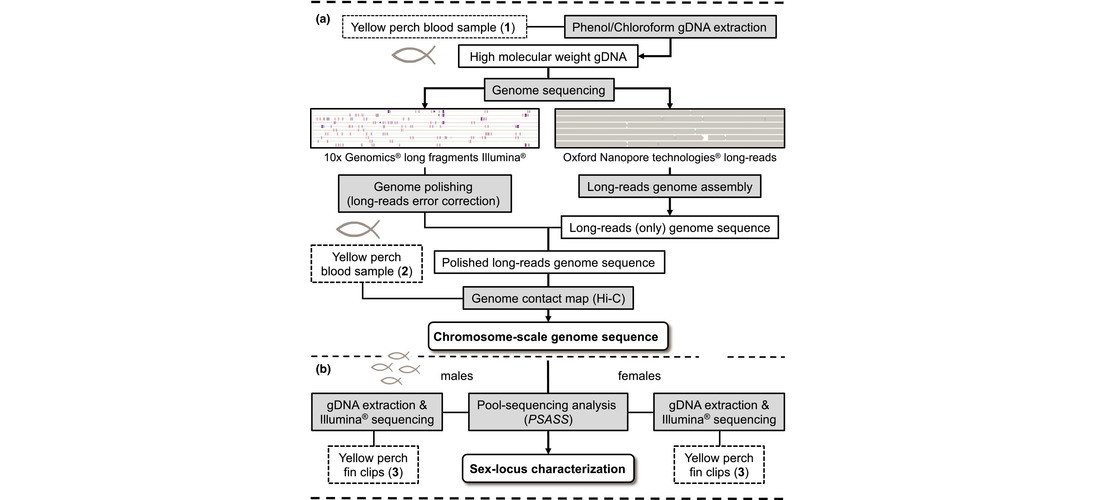
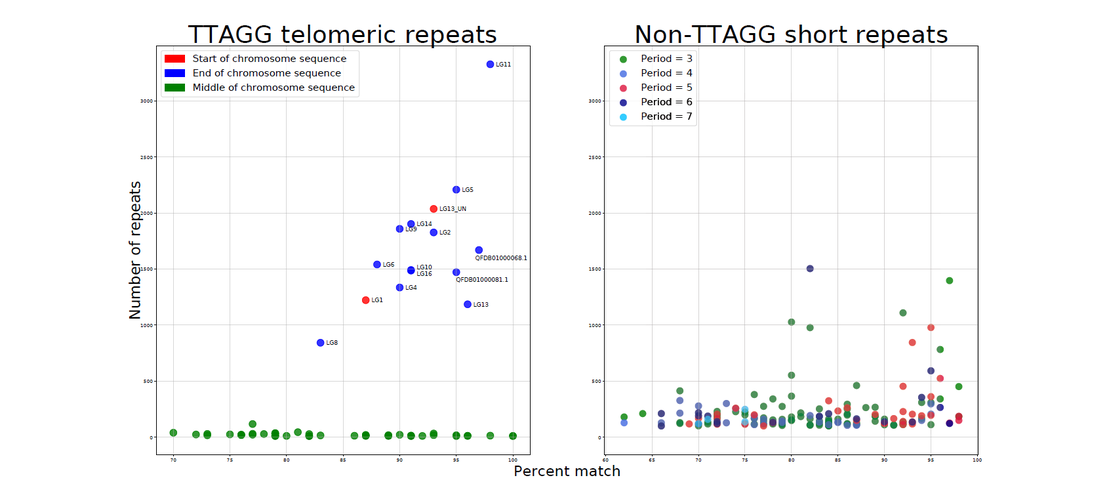
the reference which do not match with the alignment. Contigs are then scaffolded in chromosomes using
BioNano optical maps, Hi-C ligation reads or a genetic map.
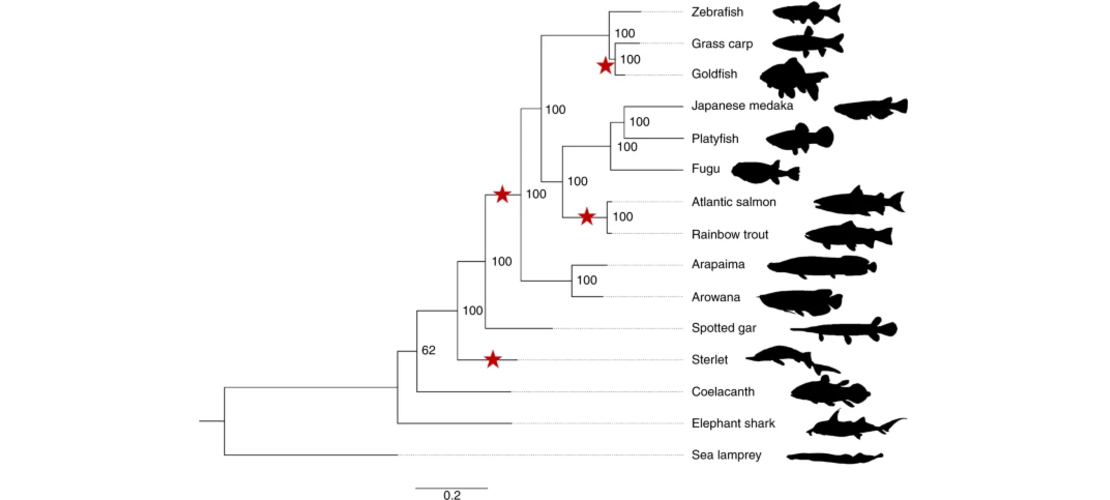
Sigenae has assembled over fifteen fish genome as well as the bee genome. Some of these assemblies
where quite complicated due to slow evolution and recent duplication as for example the sterlet
sturgeon genome (Nature Ecology & Evolution,
2020). We have taken part in the assemblies of the
SeqOccin project. And we have also been in charge of the assemblies of the
GenoFish project of which some genomes are references at the NCBI or Ensembl like the
javafish, the
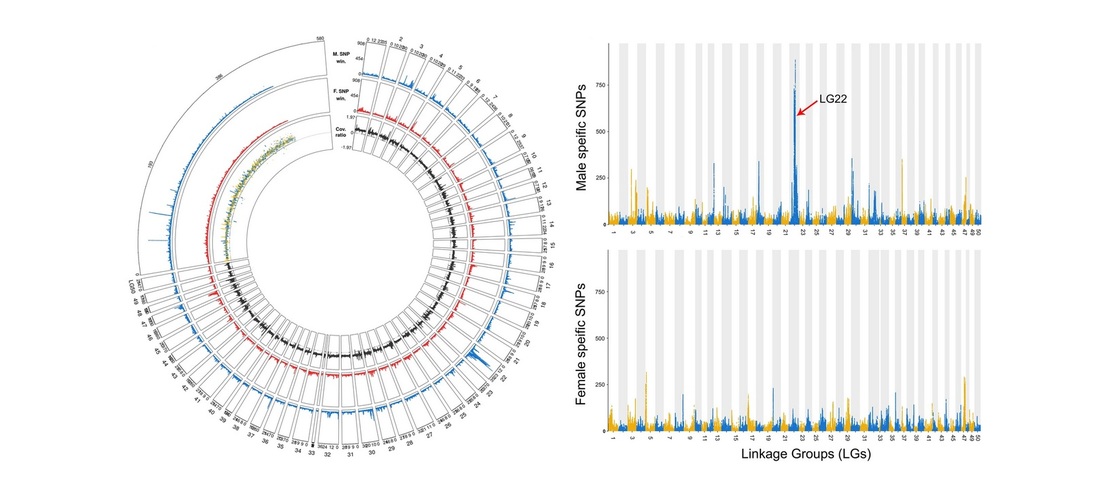
yellow perch or the
european perch.
Assembly projects recently published